利用赖氨酸和18-Crown-6之间的选择性络合高效分离甲基化肽
蛋白质甲基化是最常见和最重要的翻译后修饰之一,在表观遗传调控,信号转导和染色质代谢中起着至关重要的作用。然而,由于甲基化形式的多样性,甲基化位点与未修饰位点之间的细微差异,以及超低的丰度,从生物样品中捕获和分离甲基化肽具有极大的挑战。为此,卿光焱研究员组发展了一种基于高效液相色谱(HPLC)中18-crown-6作为流动相添加剂的分离甲基化和非甲基化肽的简单高效方法。赖氨酸和18-crown-6之间的选择性络合显着增加了肽在C18固定相上的保留力,从而导致赖氨酸甲基化和非甲基化肽之间的出色基线分离。通过核磁共振滴定,生物层干涉技术和量子化学计算验证了可能的结合机理。通过建立简单的富集方法,可以实现良好的选择性,并从含有10倍牛血清白蛋白(BSA)胰蛋白酶的复杂肽样品中,成功分离出信噪比(S / N)大大提高的四种甲基化肽。通过选择rLys N作为消化组蛋白的酶,可以根据我们的富集方法很好地识别组蛋白中的甲基化信息。这项研究将为在翻译后修饰蛋白质组学中,选择性富集赖氨酸甲基化肽提供新途径。
Highly Efficient Separation of Methylated Peptides Utilizing Selective Complexation between Lysine and 18-Crown-6
Qianying Sheng, Cunli Wang, Minbo Lan*, and Guangyan Qing*
Anal. Chem., 2020, 92, 15663.
DOI: 10.1021/acs.analchem.0c04158
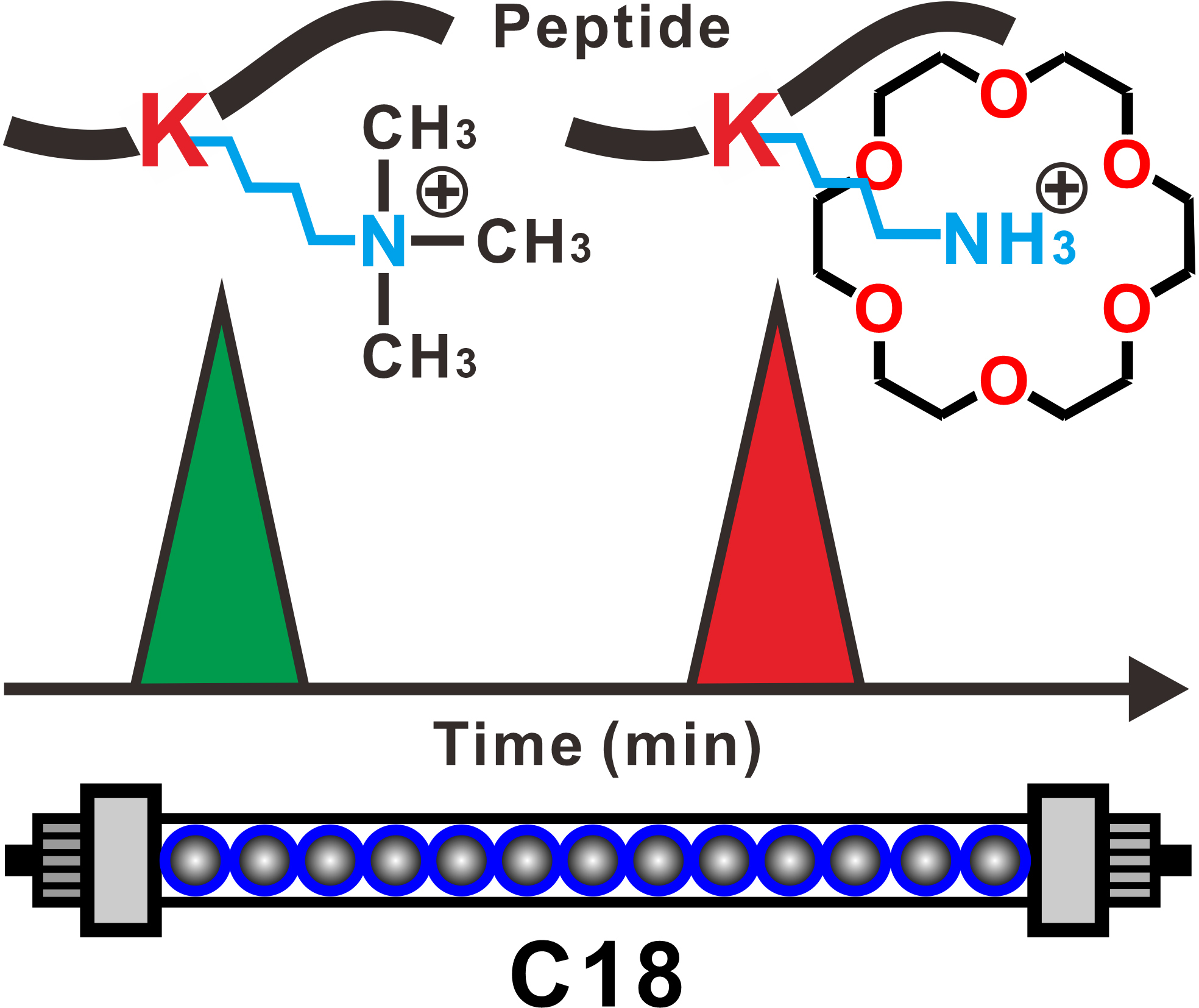
Protein methylation is one of the most common and important post-translational modifications and plays vital roles in epigenetic regulation, signal transduction and chromatin metabolism. However, due to diversity of methylation forms, slight difference between methylated sites and non-modified ones, and ultra-low abundance, it is extraordinarily challenging to capture and separate methylated peptides from biological samples. Here, we introduce a simple and highly efficient method to separate the methylated and non-methylated peptides based on using 18-crown-6 as a mobile phase additive in high-performance liquid chromatography (HPLC). Selective complexation between lysine and 18-crown-6 remarkably increases the retention of the peptides on a C18 stationary phase, leading to an excellent baseline separation between the lysine methylated and non-methylated peptides. A possible binding mechanism is verified by nuclear magnetic resonance titration, biolayer interferometry technology and quantum chemistry calculation. Through establishment of a simple enrichment methodology, a good selectivity is achieved and four methylated peptides with greatly improved signal-to-noise (S/N) ratio are successfully separated from a complex peptide sample containing 10 fold of bovine serum albumin (BSA) tryptic digests. By selecting rLys N as an enzyme to digest histone, methylation information in the histone could be well identified based on our enrichment method. This study will open an avenue and provide a novel insight for selective enrichment of lysine methylated peptides in post-translational modification proteomics.








